Sarcomas
Sarcomas Óseos
Definición
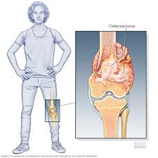
El cáncer de hueso es poco común en los adultos. Se origina en las células que conforman los huesos. El cáncer ocurre cuando las células comienzan a crecer sin control. Casi cualquier célula del cuerpo puede convertirse en cáncer y propagarse a otras partes del cuerpo. Los tumores óseos verdaderos (o primarios) se originan en el hueso en sí y se denominan sarcomas. Estos son tumores malignos, lo que significa que son cancerosos.
Los tumores óseos suelen aparecer en zonas de hueso que crecen rápidamente (Metáfisis). Podemos clasificarlos en diferentes subtipos:
- Condrosarcoma
- Condroma
- Sarcoma de Ewing
- Osteosarcoma
- Tumor de células gigantes
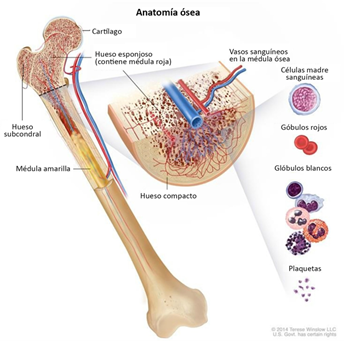
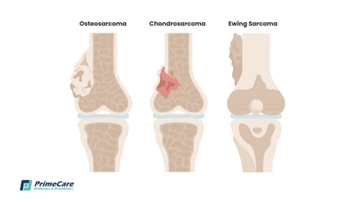
Epidemiología
Existen muchos tipos diferentes de cánceres de huesos. El nombre que se les asigna está en función de la parte de hueso o de tejido adyacente afectada y del tipo de células que forman el tumor.
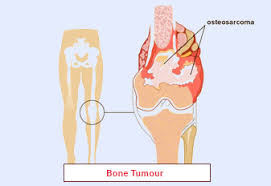
Los sarcomas óseos son poco frecuentes representando hasta el 1% de los tumores malignos. El osteosarcoma es el tipo más común frecuente de tumor primario con 2-3 nuevos casos por millón de persona cada año. La mayoría de las veces afecta a personas jóvenes de entre 10 y 30 años de edad
El condrosarcoma se origina en las células del cartílago, y es el segundo cáncer de hueso primario más común. Es poco común ver este cáncer en personas menores de 20 años de edad
El tumor de Ewing es el tercer tipo de cáncer de hueso primario más común, y el segundo tipo más común en niños, adolescentes y adultos jóvenes. La mayoría de los tumores de Ewing se desarrollan en los huesos, pero pueden originarse en otros tejidos y órganos. Los sitios en los que este cáncer se desarrolla más comúnmente son la pelvis, la pared torácica (por ejemplo, las costillas o los omóplatos), y los huesos largos de las piernas o los brazos.
Factores de riesgo

Actualmente no está claro por qué se producen los sarcomas óseos. Se han identificado algunos factores de riesgo. Un factor de riesgo aumenta el riesgo de aparición de cáncer, pero no es ni necesario ni suficiente para causar cáncer. Un factor de riesgo no es una causa en sí mismo.
Algunas personas con estos factores de riesgo* nunca desarrollarán un sarcoma óseo y otras personas sin ninguno de estos factores de riesgo sin embargo pueden desarrollar un sarcoma óseo.
- Sd de Li- Fraumeni
- Retinoblastoma Hereditario
- Enfermedad ósea de Paget
- Radiación Ionizante
Cuadro Clínico
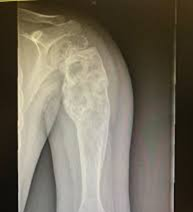
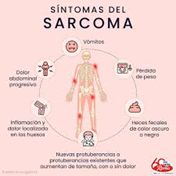
Los sarcomas óseos pueden ser asintomáticos durante un largo tiempo, y la inflamación (hinchazón y enrojecimiento) sólo estará presente si el tumor ha progresado a través del hueso cortical. Los síntomas dependen del tamaño y la localización del tumor.
El dolor de huesos es el síntoma más común: dolor que progresa gradualmente hasta convertirse en un dolor persistente.
El tumor también puede debilitar los huesos, causando fracturas espontáneas o fracturas después de una lesión menor o una caída.
Pueden estar presentes problemas de nervios debido a la constricción de los nervios ocasionada por el tumor. Los sarcomas óseos también pueden detectarse por casualidad durante una investigación de otros síntomas o durante una operación de rutina.
Diagnóstico
El diagnóstico de los sarcomas óseos se basa en los siguientes exámenes: Historial médico y exámenes clínicos
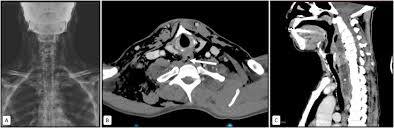
Examen radiológico.
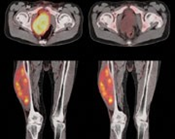
Se usa una amplia gama de técnicas de adquisición de imágenes para poder ver dentro del cuerpo y determinar el alcance de un sarcoma óseo y establecer la presencia o ausencia de enfermedad metastásica distante.
- Rayos X
- Resonancia magnética
- TAC contrastado
- Gammagrafía Ósea
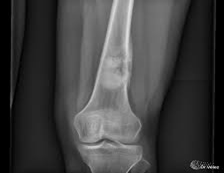
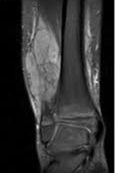
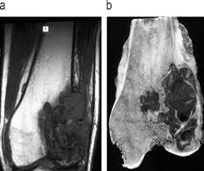
Examen histopatológico
El examen histopatológico, es decir, el examen de tejidos en un microscopio, se lleva a cabo sobre una biopsia o porción de tejido tras la extirpación de todo el tumor por cirugía. Sólo la evaluación histopatológica del tumor revelará si el tumor es un sarcoma óseo y, en su caso, de qué tipo. También proporcionará el “grado de malignidad”, esto es, una clasificación de la agresividad de las células del cáncer.
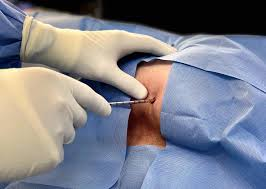
Tratamiento

Los médicos usan la estadificación* (clasificación de los tumores por etapas) para determinar la extensión del tumor en el cuerpo, lo que resulta un indicador importante del pronóstico*
La carga tumoral y la presencia de enfermedad distante detectable son los dos principales factores que se toman en consideración en la estadificación* clínica de estas enfermedades.
La determinación del estadio es fundamental para tomar la decisión correcta acerca de qué tratamiento usar. Cuanto más bajo sea el estadio, mejor será el pronóstico
El tratamiento generalmente combinará terapias que:
Afecten al tumor localmente, como cirugía o radioterapia*
Afecten a las células tumorales presentes en otros lugares del cuerpo a través de terapia sistémica, como la quimioterapia*
La elección del tratamiento dependerá del tipo y el estadio del tumor y también tendrá en consideración el riesgo para el paciente.

Plan de tratamiento para enfermedades localizadas
Los sarcomas óseos están localizados cuando están confinados en su sitio primario* y no se han extendido a los tejidos cercanos ni a otras regiones del cuerpo. En este estadio, el principal objetivo terapéutico es extirpar todo el tumor por cirugía siempre que sea posible. La radioterapia* y la quimioterapia* también pueden usarse para aumentar las posibilidades de cura definitiva o reducir el riesgo de que el tumor vuelva a aparecer.
Cirugía
Con mucha frecuencia, la cirugía es el método de tratamiento estándar usado para el sarcoma óseo localizado. Dado que los sarcomas óseos son poco comunes, la cirugía debería ser realizada por un cirujano que esté especializado en tratar este tipo de tumor.
Radioterapia*
En los sarcomas óseos, la radioterapia puede usarse antes (neoadyuvante) de la cirugía (para reducir el tamaño del tumor y permitir que pueda extirparse completamente) o después (adyuvante) de la cirugía (para acabar con todas las células tumorales restantes.

La quimioterapia puede considerarse aisladamente o en combinación con la radioterapia, y puede administrarse antes o después de la cirugía en los casos de enfermedad localizada. Se considera especialmente en estas 2 situaciones:
En el caso del tratamiento del osteosarcoma la quimioterapia tiene un papel muy bien establecido en la prevención de recaídas locales y distantes, y por lo general se administra de forma tanto preoperatoria como postoperatoria por un período acumulativo de 6-10 meses. En el sarcoma de Ewing, la quimioterapia generalmente se administra cada tres semanas, de forma preoperatoria y posoperatoria, durante alrededor de 10-12 meses, con pautas que incluyen al menos 5-6 fármacos diferentes. Puede ser utilizada en combinación con radioterapia.


Referencias
- Bone Cancer NCCN Guidelines for patients 2024 Version 1.2024
- Bone Sarcomas: A guide for patients ESMO Clinical Practice Guidelines
- American Cancer Society: Bone Sarcoma
SARCOMAS DE TEJIDOS BLANDOS
Definición
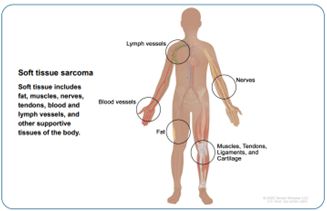
El cáncer se origina cuando las células comienzan a crecer sin control. Las células de casi cualquier parte del cuerpo pueden volverse cancerosas y propagarse a otras áreas. Un sarcoma es un tipo de cáncer que comienza en tejidos como el hueso o el músculo.
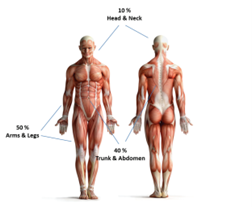
Los sarcomas de tejidos blandos pueden desarrollarse en tejidos blandos como la grasa, los músculos, los nervios, los tejidos fibrosos, los vasos sanguíneos o los tejidos profundos de la piel. Se pueden encontrar en cualquier parte del cuerpo. Las partes del cuerpo más afectados son piernas 50%, seguidos de tórax y abdomen 40% y cabeza y cuello 10%
Epidemiología
Los sarcomas de tejidos blandos son tumores raros, el riesgo de por vida de presentar un sarcoma de tejidos blandos es del 0.15 - 0.50%. El pico de incidencia es a la edad de 50-60 años. Las estimaciones de la Sociedad Estadounidense del cáncer para los sarcomas de tejidos blandos en los Estados Unidos para 2025 son:
- Se diagnosticarán aproximadamente 13.520 nuevos sarcomas de tejidos blandos (7.600 en hombres y 5.920 en mujeres).
- Se estima que alrededor de 5.420 personas (2.960 hombres y 2.450 mujeres) morirán de sarcomas de tejidos blandos.
Estas estadísticas incluyen tanto a adultos como a niños.
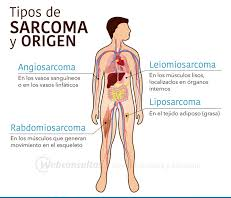
Los tipos más comunes de sarcoma en adultos son:
- Sarcoma pleomórfico indiferenciado (anteriormente llamado histiocitoma fibroso maligno)
- Liposarcoma
- Leiomiosarcoma
Factores de riesgo y genética
Un factor de riesgo es cualquier cosa que cambie la probabilidad de padecer una enfermedad como el cáncer. Cada tipo de cáncer tiene diferentes factores de riesgo. Tener un factor de riesgo, o incluso muchos, no significa que vayas a padecer cáncer. Muchas personas padecen cáncer sin tener un factor de riesgo.
No está claro porqué ocurren los sarcomas de tejidos blandos sin embargo se han identificado diferentes factores de riesgo que mayoritariamente tienen que ver con alteraciones genéticas
- Retinoblastoma
- Neurofibromatosis
- Síndrome de Li-Fraumeni
- Síndrome de Lynch
- Síndrome de Poliposis adenomatosa familiar
Cuadro Clínico
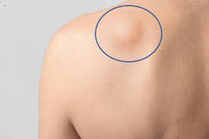
Aproximadamente la mitad de los sarcomas de tejidos blandos comienzan en un brazo o una pierna. La mayoría de las personas notan una masa o bulto que crece con el tiempo (semanas o meses). La masa puede doler o no.
Cuando los sarcomas crecen en la parte posterior del abdomen (el retroperitoneo), los síntomas suelen deberse a otros problemas que causa el tumor. Por ejemplo, pueden causar obstrucción o sangrado del estómago o los intestinos. Pueden presionar nervios, vasos sanguíneos u órganos cercanos. Pueden crecer lo suficiente como para que el tumor se sienta en el abdomen. A veces, los tumores causan dolor. Aproximadamente 4 de cada 10 sarcomas comienzan en el abdomen.
Diagnóstico
Si tiene signos o síntomas que sugieren que podría tener un sarcoma de tejidos blandos, es probable que el médico necesite realizarle pruebas para determinar si tiene cáncer.
Historial médico y examen físico
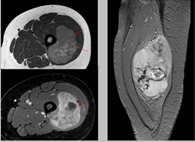
Pruebas de imágen
Las pruebas de diagnóstico por imágenes utilizan ondas sonoras, rayos X, campos magnéticos o sustancias radiactivas para crear imágenes del interior del cuerpo. Las pruebas de diagnóstico por imágenes pueden realizarse por diversos motivos, como:
- Para observar áreas sospechosas que podrían ser cáncer,
- Para ver si el cáncer se ha propagado y hasta qué punto
- Para ayudar a determinar si el tratamiento está funcionando
Las imágenes más representativas o más comunes son:
- Radiografías de tórax
- Ultrasonido
- Tomografía Computarizada
- PET scan
- Resonancia Magnética
Biopsia
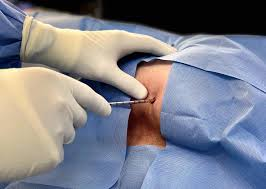
Si se sospecha que se trata de un sarcoma de tejidos blandos a partir de exámenes y pruebas de diagnóstico por imágenes, es necesario realizar una biopsia para saber con certeza que se trata de un sarcoma y no de otro tipo de cáncer o de una enfermedad benigna (no cancerosa).
En una biopsia, el médico extrae una pequeña porción del tumor. Este tejido se examina con un microscopio y también se pueden realizar otras pruebas de laboratorio.
Tratamiento
Dependiendo del tipo de sarcoma, además de la etapa clínica se utilizan diferentes tipos de tratamiento.
La cirugía se utiliza comúnmente para tratar los sarcomas de tejidos blandos. Según el sitio y el tamaño del sarcoma, la cirugía podría ser capaz de extirpar el cáncer. El objetivo de la cirugía es extirpar todo el tumor junto con al menos 1 a 2 cm (menos de una pulgada) del tejido normal que lo rodea. El estándar es la cirugía para extirpar el tumor sin amputación. Esto se llama cirugía para preservar la extremidad. Puede ir seguido de radioterapia.
En ocasiones, se puede administrar quimioterapia (quimio), radiación o ambas antes de la cirugía. Esto se denomina tratamiento neoadyuvante. Se puede utilizar para reducir el tamaño del tumor y poder extirparlo por completo. También se puede administrar quimioterapia o radiación antes de la cirugía para tratar sarcomas de alto grado cuando existe un alto riesgo de propagación del cáncer.
También se puede utilizar quimioterapia y/o radioterapia después de la cirugía. Esto se denomina tratamiento adyuvante. El objetivo es destruir cualquier célula cancerosa que pueda quedar en el cuerpo para reducir el riesgo de que el cáncer regrese.
La quimioterapia (quimio) es el uso de medicamentos que se administran por vía intravenosa o por vía oral para tratar el cáncer. Estos medicamentos ingresan al torrente sanguíneo y llegan a todas las áreas del cuerpo, lo que hace que este tratamiento sea útil para el cáncer que se ha propagado (ha hecho metástasis) a otros órganos. Según el tipo y el estadio del sarcoma, la quimioterapia se puede administrar como tratamiento principal o como tratamiento adyuvante (adición) a la cirugía. Diferentes tipos de sarcoma responden mejor a la quimioterapia que otros y también responden a diferentes tipos de quimioterapia. La quimioterapia para el sarcoma de tejidos blandos generalmente utiliza una combinación de varios medicamentos contra el cáncer.
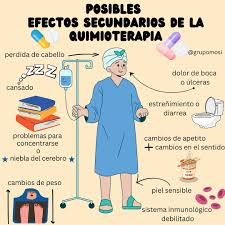
Los medicamentos de quimioterapia matan las células cancerosas, pero también dañan algunas células normales. Esto provoca efectos secundarios. Los efectos secundarios dependen del tipo de medicamento, la cantidad que se toma y la duración del tratamiento. Los efectos secundarios más comunes de la quimioterapia incluyen:
- Náuseas y vómitos
- Pérdida de apetito
- Pérdida de cabello
- Llagas en la boca
- Fatiga
- Recuentos sanguíneos bajos


Inmuno Terapia
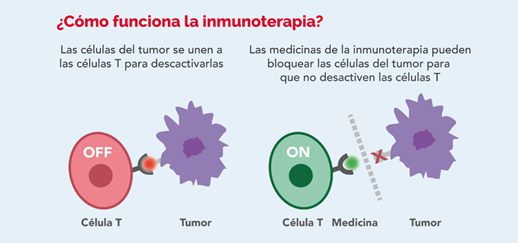
La inmunoterapia es el uso de medicamentos para ayudar al sistema inmunológico de una persona a reconocer y destruir las células cancerosas de manera más efectiva
Una parte importante del sistema inmunitario es su capacidad de evitar atacar a las células normales del cuerpo. Para ello, utiliza proteínas de “punto de control” en las células inmunitarias u otras células que deben activarse (o desactivarse) para iniciar una respuesta inmunitaria. Las células cancerosas a veces utilizan estos puntos de control para evitar ser atacadas por el sistema inmunitario. Pero los medicamentos que se dirigen a estos puntos de control, conocidos como inhibidores de puntos de control, se pueden utilizar para tratar a algunas personas con sarcomas de tejidos blandos.
Referencias
- Soft Tissue Sarcomas NCCN Guidelines for patients 2024 Version 1.2024
- Soft Tissue Sarcomas: A guide for patients ESMO Clinical Practice Guidelines
- American Cancer Society: Soft Tissue Sarcoma